

A Man with a Recurrent Left Pleural Effusion
In late 2020, a new disease entity that was referred to as VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) syndrome was reported.
Q: What is the cause of VEXAS syndrome?
A: VEXAS syndrome is caused by somatic mutations in UBA1 (the gene that encodes ubiquitin-like modifier activating enzyme 1), which initiates the first step of cellular ubiquitylation. The mutations arise later in life and are confined to hematopoietic precursor cells in bone marrow and myeloid cells (e.g., neutrophils and monocytes) in peripheral blood. Since UBA1 is an X-linked gene and women are protected by a second allele, VEXAS syndrome occurs almost exclusively in men.
Q: What are some of the diseases that VEXAS syndrome can mimic?
A: Patients with VEXAS syndrome can have inflammatory symptoms that mimic rheumatologic diseases. Specifically, patients with VEXAS syndrome can meet diagnostic or classification criteria for one or more diseases, including relapsing polychondritis, polyarteritis nodosa, giant-cell arteritis, Sweet’s syndrome, ANCA-associated vasculitis, systemic lupus erythematosus, and adult-onset Still’s disease.
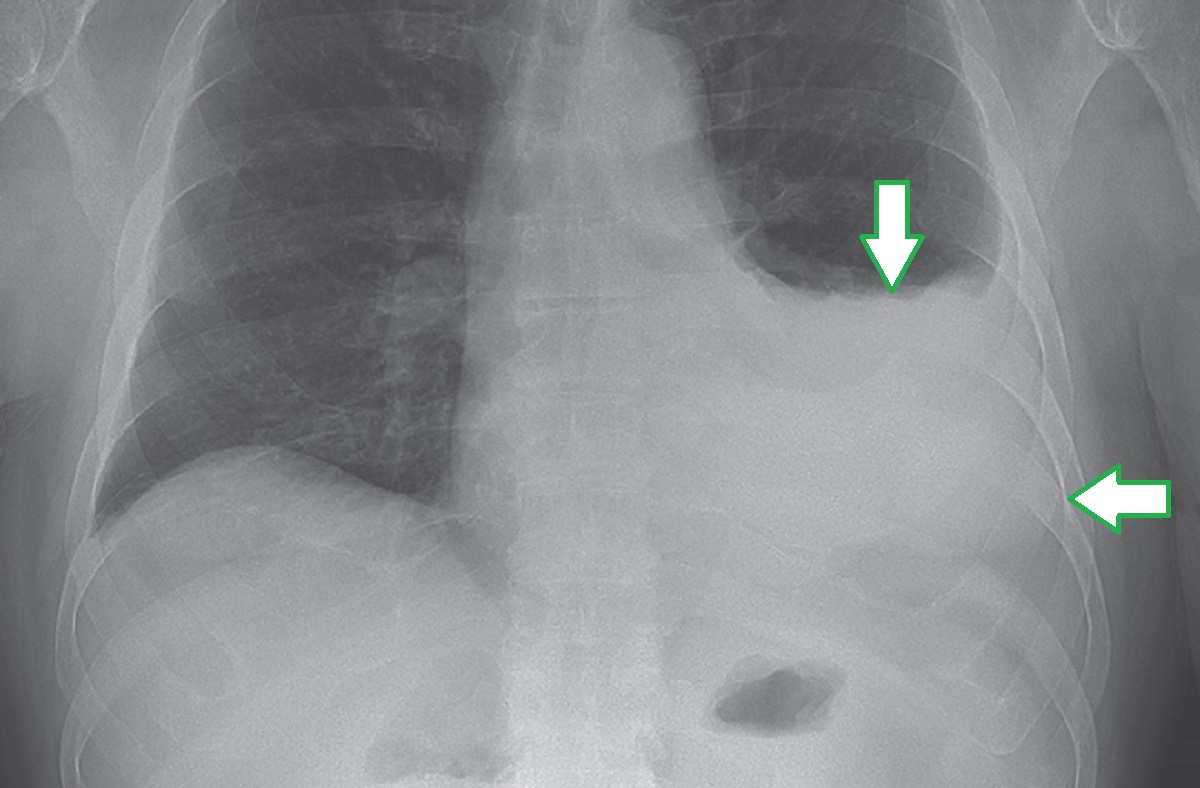
Q: Describe common clinical features of VEXAS syndrome.
A: Chondritis is a common clinical feature in patients with VEXAS syndrome, is typically confined to the ear or nose, and can be transient and subtle without resultant damage. Fever and cutaneous manifestations that reflect neutrophilic dermatosis, leukocytoclastic vasculitis, or medium-vessel vasculitis are common in patients with VEXAS syndrome. Pulmonary involvement can occur, often in the later stages of disease. Sterile neutrophilic alveolitis is the most common pulmonary manifestation, but exudative pleural effusions have been reported in a minority of patients. Musculoskeletal conditions that can mimic polymyalgia rheumatica have been described. Recurrent thromboembolic disease, in the absence of traditional risk factors for hypercoagulability, is common in patients with VEXAS syndrome. Inflammation can be severe and difficult to treat, and only glucocorticoids have shown consistent effectiveness in ameliorating symptoms.
Q: What are some of the hematologic manifestations of VEXAS syndrome?
A: In addition to systemic inflammatory disease, progressive bone marrow failure is a defining feature of VEXAS syndrome. Mutations in UBA1 in bone marrow stem cells and in hematopoietic progenitor cells, with subsequent clonal expansion, result in hematologic abnormalities. Macrocytosis is the most common hematologic manifestation and is typically an early feature of disease. Progressive cytopenias, especially thrombocytopenia and lymphopenia, occur in the later phases of disease. Numerous cytoplasmic vacuoles in erythroid and myeloid precursor cells are present in the bone marrow aspirate in almost all patients with VEXAS syndrome. The presence of cytoplasmic vacuoles is not a specific finding; they can also be seen in marrow elements in association with zinc toxicity, copper deficiency, alcohol use, and myelodysplastic syndrome, as well as in certain types of neoplastic cells. However, a mutation in UBA1 indicates that the vacuoles are related to VEXAS syndrome and not to some other cause. Genetic testing for VEXAS syndrome may be performed on either bone marrow or peripheral-blood cells, since mutational burden is typically high in both compartments.
Read the NEJM Case Records of the Massachusetts General Hospital here.
Source: NEJM Resident 360