

GTC posters at 67th ASH® Annual Meeting (2025)
We are delighted to share that GTC collaborated on 12 abstracts at the ASH 2025 Annual Meeting in Orlando, December 6-9. The work represents novel data across machine-learning prediction models, transcriptomic signatures, cell-free RNA (cfRNA) analytics, ethnic-ancestry outcomes, high risk multiple myeloma features and more – underscoring GTC’s commitment to innovation in precision hematology.
Here are the four posters GTC presented independently

Bone Marrow Microenvironment Overlap Between Vexas And Myelodysplastic Syndrome Demonstrated By Targeted Transcriptomic And Artificial Intelligence

Developing Artificial Intelligence-Based Transcriptomic Signature For The Diagnosis Of Dark Zon Lymphoma In Patients Without Myc Gene Rearrangement
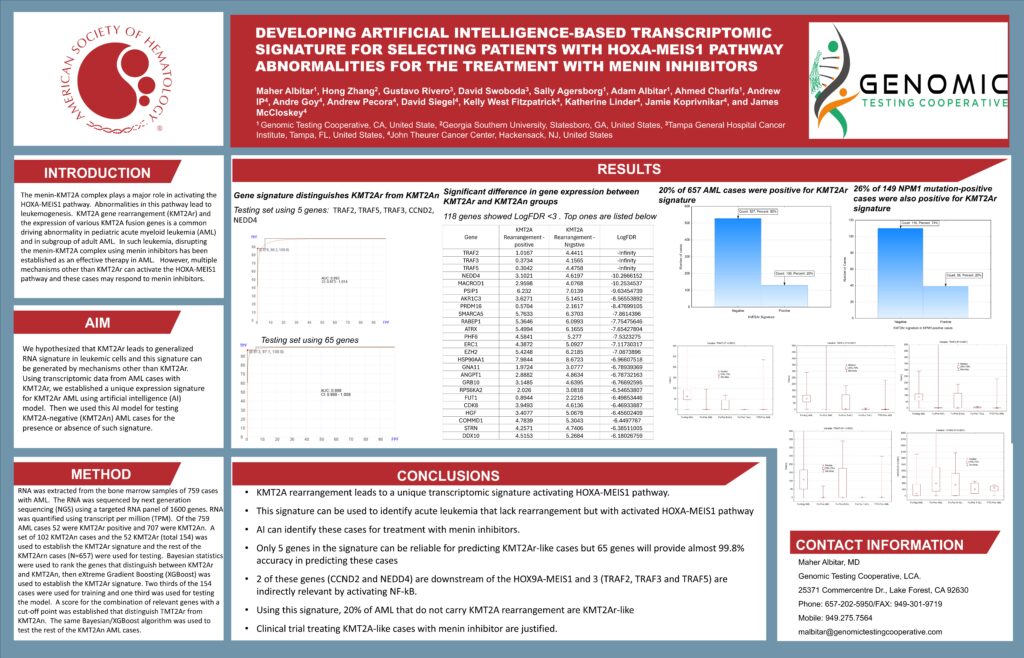
Developing Artificial Intelligence-Based Transcriptomic Signature For Selecting Patients With Hoxa-Meis1 Pathway Abnormalities For The Treatment With Menin Inhibitors
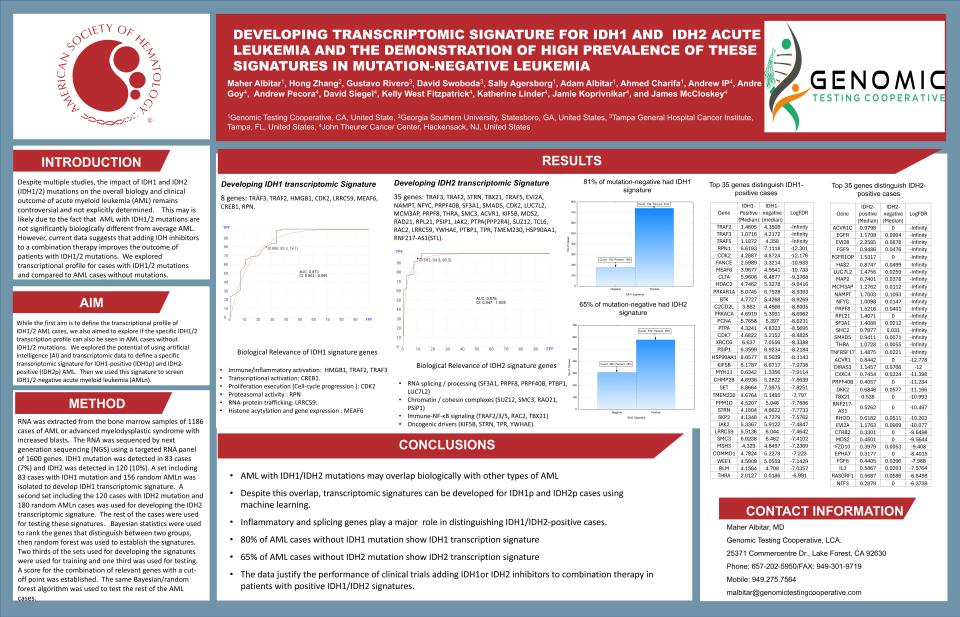
Developing Transcriptomic Signature For Idh1 And Idh2 Acute Leukemia And The Demonstration Of High Prevalence Of These Signatures In Mutation-Negative Leukemia
Here is the full list of abstracts and our collaborators
Abstract Number: abs25-2858
Title: Liquid-biopsy mutation landscape and its concordance with skin biopsies in cutaneous T-cell lymphoma
Room: OCCC – Tangerine Ballroom F2
Date: Sunday, December 7, 12:30 PM – 12:45 PM EST
Part of session 625. T Cell, NK Cell, or NK/T Cell Lymphomas: Clinical and Epidemiological: Fresh insight for Cutaneous and Virus-associated TCLs
Authors:
Yoni Sacknovitz¹, Maher Albitar², Sara Suhl¹, Alexander Kaminsky¹, Brigit Lapolla³, Joshua Kent³, Maggie Zhou¹, Seda Tolu⁴, Alejandro Gru⁵, Barbara Pro⁴, Tatyana Feldman⁶, Larisa Geskin³
Affiliations:
¹ Columbia University Vagelos College of Physicians and Surgeons, New York, NY, United States
² Genomic Testing Cooperative, Lake Forest, CA, United States
³ Columbia University Irving Medical Center, Dermatology, New York, NY, United States
⁴ Columbia University Irving Medical Center, Hematology-Oncology, New York, NY, United States
⁵ Columbia University Irving Medical Center, Dermatopathology, New York, NY, United States
⁶ Hackensack Meridian Health, Hackensack, NJ, United States
Abstract Body:
Liquid biopsy (LBx) is widely utilized for clinical evaluation and therapeutic monitoring in solid and hematologic malignancies (Talotta et al., Frontiers in Oncology 2023: https://doi.org/10.3389/fonc.2023.1295958; Huang et al., Molecular Cancer 2024: https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-024-01909-6). Analyzing circulating cell-free DNA (cfDNA) and RNA (cfRNA) through LBx can offer minimally invasive insights into tumor burden and disease dissemination (Huang et al. 2024). Diagnosing cutaneous T-cell lymphoma (CTCL) is challenging both clinically and histologically. LBx as a diagnostic tool could support detection of disease and potentially reduce reliance on skin biopsies.
LBx interpretation in CTCL is complicated by overlapping somatic mutations commonly detected in clonal hematopoiesis of indeterminate potential (CHIP), which are also present in other hematologic malignancies (Marnell et al., Journal of Molecular and Cellular Cardiology 2021: https://doi.org/10.1016/j.yjmcc.2021.06.003). CHIP-associated mutations in genes such as TET2 and DNMT3A are shared with myeloid and T-cell neoplasms, adding complexity (Kusne et al., Leukemia Research 2022: https://doi.org/10.1016/j.leukres.2022.106907).
Mutations in CTCL commonly involve pathways regulating T-cell receptor signaling, cytokine activation, cell-cycle progression, apoptosis, and epigenetic control. Epigenetic regulators — including TET2, DNMT3A, ASXL1, ARID1A, and genes encoding histone-modifying enzymes — are frequently mutated in both CTCL and myeloid neoplasms (Choi et al., Nature Genetics 2015: https://doi.org/10.1038/ng.3335).
In this study, cfDNA and cfRNA from CTCL patients were evaluated, including comparisons between LBx findings, skin biopsy (SBx) results, and paired LBx–SBx samples. The objective is to characterize LBx-detected mutations associated with CTCL to support more accurate LBx interpretation and to reinforce its potential diagnostic value in CTCL.
Abstract Number: abs25-14588
Title: AI-derived prediction of response and relapse to venetoclax plus hypomethylating agent–based therapy in acute myeloid leukemia
Room: OCCC – West Halls B3-B4
Date: Saturday, December 6, 05:30 PM – 07:30 PM EST
Part of session 618. Acute Myeloid Leukemias: Biomarkers and Molecular Markers in Diagnosis and Prognosis: Poster I
Authors:
Sarvarinder Gill¹, Maher Albitar², Jamie Koprivnikar¹, Katherine Linder¹, Ayrton Bangolo¹, Behzad Amoozgar¹, Lili Zhang¹, James McCloskey¹
Affiliations:
¹ John Theurer Cancer Center at Hackensack University Medical Center, Hackensack, NJ, United States
² Genomic Testing Cooperative, Lake Forest, CA, United States (corrected city for GTC corporate HQ — Lake Forest, CA)
Abstract Body:
Venetoclax combined with hypomethylating agents (Ven-HMA) has become a standard of care for older or unfit patients with acute myeloid leukemia (AML). Despite high initial response rates, many patients experience primary refractory disease or later relapse, which remain major clinical challenges (DiNardo et al., NEJM 2020: https://doi.org/10.1056/NEJMoa1911361). Existing risk stratification approaches, such as the ELN classification system, were primarily designed around outcomes with intensive chemotherapy and show limited predictive value for Ven-HMA–based regimens (Döhner et al., Blood 2022: https://doi.org/10.1182/blood.2022016867).
There is an unmet need for robust and validated biomarkers that can guide individualized treatment decisions and predict treatment failure. Artificial intelligence (AI) models can integrate multi-modal datasets to identify complex patterns that are not apparent through conventional analysis. In this study, AI-driven predictive models were developed and validated using comprehensive clinical, genomic, and transcriptomic data from 128 patients with AML or high-risk myelodysplastic syndrome (MDS) treated with Ven-HMA therapy. The purpose of this work is to improve prediction of therapeutic response and relapse risk, supporting personalized treatment strategies in AML.
Abstract Number: abs25-14645
Title: Integrative clinical and molecular analysis of outcome in elderly African ancestry acute myeloid leukemia
B- and T-cell clonality using peripheral blood cell-free RNA (cfRNA) in liquid biopsy
Room: OCCC – West Halls B3-B4
Date: Saturday, December 6, 05:30 PM – 07:30 PM EST
Part of session 618. Acute Myeloid Leukemias: Biomarkers and Molecular Markers in Diagnosis and Prognosis: Poster I
Authors:
Jonathan Andreadakis¹, Olivia Wilkins², Gary Kupfer², Alexander Shkembi¹, Amy Wu¹, John Basile¹, Anna Krzeczowska³, Alexandra Thalberg⁴, Linda Linderbeck³, Nidia Zapata⁵, Marci O’Driscoll³, Suzane Silbert³, Anne Renteria⁶, Maher Albitar⁷, Jason Salemi¹, Amy Alman¹, Tiphaine Martin⁸, Martha Mims⁹, David Swoboda³, Gustavo Rivero³, Lacey Williams¹⁰
Affiliations:
¹ University of South Florida, Tampa, FL, United States
² Georgetown University, Washington, DC, United States
³ Tampa General Hospital Cancer Institute, Tampa, FL, United States
⁴ University of Minnesota, Minneapolis, MN, United States
⁵ Instituto de Cancerología de la Ciudad de México, Mexico City, Mexico
⁶ New York University, Mineola, NY, United States
⁷ Genomic Testing Cooperative, Lake Forest, CA, United States
⁸ Icahn School of Medicine at Mount Sinai, New York, NY, United States
⁹ Baylor College of Medicine, Houston, TX, United States
¹⁰ University of North Carolina, Hematology, Chapel Hill, NC, United States
Abstract Body:
African ancestry (AA) has been associated with inferior overall survival in acute myeloid leukemia (AML), as reported in SEER registry analyses and CALGB-Alliance studies (Kirtane et al., Blood Advances 2021: https://doi.org/10.1182/bloodadvances.2021005398). Prior work has shown that elderly AA AML is characterized by increased myelodysplasia-related chromosomal abnormalities (e.g., 7q, −7) and DNA repair mutations such as RAD21 and FBXW7, along with social vulnerability factors influencing outcomes [Williams et al., BMC Cancer 2025 — in press; reference available upon publication].
AML blasts typically display myeloid markers reflecting normal hematopoiesis but accumulate key genomic abnormalities at varying stages of differentiation arrest. We hypothesized that relationships among genomic alterations, maturation state, and transcriptomic dysregulation would better explain AA-specific leukemogenesis.
After IRB approval, data from 72 AA and 169 non-AA elderly AML patients aged ≥60 years were analyzed. Descriptive statistics summarized clinical and molecular features. Chi-square and Fisher exact tests evaluated categorical data. Survival was assessed by Kaplan–Meier and Cox regression analyses. Differential gene expression in newly diagnosed AML (12 AA and 11 non-AA cases) was evaluated using one- and two-sample t-tests with Bonferroni correction applied. ToppGene Suite (https://toppgene.cchmc.org/) identified enriched canonical pathways. Statistical analyses were performed using SAS version 9.4.
Median age was 73 years (61–93) in AA and 71 years (61–90) in non-AA patients; 65% were male. In AA, ELN 2022 stratification was 1/60 (1.6%) favorable, 18/60 (30%) intermediate, and 41/60 (68.3%) adverse risk, compared with 13/136 (10%), 52/136 (38.2%), and 71/136 (52.2%) in non-AA AML (p=0.04). ELN 2022 category (HR=0.18; 95% CI 0.09–0.42; p<.0001) and age (HR=1.02; 95% CI 1.01–1.03; p=0.0006) were independently associated with OS.
Compared with non-AA AML, AA AML demonstrated absent core-binding factor abnormalities (0/21 vs 9/35; p=0.01), lower NPM1 mutation frequency (1/27 vs 12/49; p=0.02), and absent FLT3-ITD mutations (0/27 vs 7/47; p=0.03), suggesting a less proliferative disease biology. Consistent with this, white blood cell counts (18.5 vs 52.8 K/µL; p=0.001) and marrow blasts (37.6% vs 57.7%; p=0.004) were lower in AA versus non-AA AML.
In flow-based phenotyping, hematopoietic progenitor cell–like (HPC-like) disease was more common in AA vs non-AA AML (75% vs 51%; p=0.03). Of 1,495 genes analyzed, 39 were significantly deregulated. Downregulated genes included FAS (p=0.0001), CD28 (p=0.006), and TNFAIP3 (p=0.01). Upregulated genes included IL13RA2 (p=0.002), BDNF (p=0.004), PRPF8 (corrected gene name; p=0.006), and ALDH1A1 (p=0.03). Pathway analysis revealed disruption of T-cell receptor signaling (FDR=1.32E-3), nucleotide excision repair (FDR=3.16E-3), and B-cell dysfunction (FDR=3.16E-3), while ligand-gated receptor signaling (FDR=2.21E-2) and protein–RNA complex assembly (FDR=4.04E-2) were enriched through upregulated genes.
Conclusions:
Elderly AA AML displays lower proliferative potential, fewer favorable-risk mutations, and a predominance of less differentiated immunophenotypes. Transcriptomic profiling indicates T- and B-cell adaptive immune dysfunction, impaired DNA repair, and increased stem-like pathways, including ALDH1A1 upregulation. Larger AA AML transcriptomic cohorts and single-cell multi-omics are needed to refine mechanistic understanding and therapeutic targeting in this at-risk population.
Keywords: Clinical Practice (Health Services and Quality), Research
Abstract Number: abs25-7865
Title: B- and T-cell clonality using peripheral blood cell-free RNA (cfRNA) in liquid biopsy
Room: OCCC – West Halls B3-B4
Date: Saturday, December 6, 05:30 PM – 07:30 PM EST
Part of session 621. Lymphomas: Translational – Molecular and Genetic: Poster I
Authors:
Adam Albitar¹, Alfonso Lam¹, Sally Agersborg¹, Ahmad Charifa¹, Pooja Phull², Noa Biran², David Vesole², Harsh Parmar², Andrew Pecora², Andrew Ip², Andre Goy², David Siegel², Maher Albitar¹
Affiliations:
¹ Genomic Testing Cooperative, Lake Forest, CA, United States
² John Theurer Cancer Center, Hackensack, NJ, United States
Abstract Body:
Cell-free RNA (cfRNA)–based liquid biopsy (LBx) is emerging as a complementary analytical approach that enhances and expands the diagnostic capabilities of LBx. While cfDNA testing has been used to assess B- and T-cell clonality in lymphoid neoplasms, sensitivity may be limited in settings with low circulating cfDNA. Because lymphoid malignancies express high levels of immunoglobulins (Igs) and T-cell receptors (TCRs), we evaluated cfRNA for clonality detection.
Using hybrid-capture next-generation sequencing (NGS) of cfRNA, B- and T-cell clonotypes and transcript expression levels were assessed in 925 LBx samples, including: 671 cases classified as normal or with low-level clonal hematopoiesis of indeterminate potential (CHIP), 449 confirmed B-cell or plasma cell neoplasms, 51 confirmed T-cell neoplasms, 327 confirmed myeloid neoplasms, and 98 solid tumors. To define clonotype expression thresholds, RNA from 550 clonal lymphoid tissue samples and 188 non-clonal tissue samples was analyzed. Tissue clonality was confirmed using flow cytometry or PCR-based assays. The RNA panel targets 1,600 genes including all TCR chains and heavy and light chain Igs. Clonotype mapping and assembly were performed using MiXCR software (https://github.com/milaboratory/mixcr).
A ≥10-fold expression difference between the top clonotype and the second-ranked clonotype was established as a stringent cutoff for confirming clonality. With clonotype-naïve analysis, B-cell clonality was detected in 36% of 449 cfRNA samples with B-cell neoplasms, 7% of normal/CHIP samples, 8% of T-cell neoplasms, 13% of myeloid neoplasms, and 14% of solid tumors. T-cell clonality, assessed through TCR-beta or TCR-gamma, was detected in 22% of T-cell neoplasms, 3% of normal/CHIP and B-cell neoplasm samples, 5% of myeloid neoplasms, and 7% of solid tumors. Due to limited TCR-alpha diversity, nonspecific clonality was frequently observed; however, excluding TRAV1-1 improved specificity and increased detection in T-cell neoplasms to 25%.
Keywords: Measurable Residual Disease; Technology and Procedures; Molecular Testing; Assays
Abstract Number: abs25-7950
Title: Somatic mutations and clinical outcomes in primary central nervous system lymphoma among Hispanic and non-Hispanic patients: A University of California Hematologic Malignancies Consortium (UCHMC) study
Room: OCCC – West Halls B3-B4
Date: Saturday, December 6, 05:30 PM – 07:30 PM EST
Part of session 621. Lymphomas: Translational – Molecular and Genetic: Poster I
Authors:
Haifaa Abdulhaq¹, Omar Mahmood¹, Nathan Futoran¹, Sani Bukari¹, Calvin Chen², Joseph Tuscano³, Aaron Tsumura³, Tamer Othman⁴, Elizabeth Brem⁵, Chenchen Niu⁵, Benjamin Heyman⁶, Michelle Don⁶, Azra Borogovac⁷, Matthew Wieduwilt⁸, Maher Albitar⁹
Affiliations:
¹ UCSF Fresno, Fresno, CA, United States
² Pathology Associates, Fresno, CA, United States
³ UC Davis Comprehensive Cancer Center, Sacramento, CA, United States
⁴ UCSF, San Francisco, CA, United States
⁵ UC Irvine, Irvine, CA, United States
⁶ UC San Diego (UCSD), San Diego, CA, United States
⁷ City of Hope National Medical Center, Duarte, CA, United States
⁸ Wake Forest University School of Medicine, Winston-Salem, NC, United States
⁹ Genomic Testing Cooperative, Lake Forest, CA, United States
Abstract Body:
Primary central nervous system lymphoma (PCNSL) is a rare and aggressive extranodal lymphoma with a distinct molecular profile. Recurrent MYD88 L265P mutations and genetic alterations involving CD79B, CARD11, PIM1, and biallelic CDKN2A loss are characteristic features that drive chronic B-cell receptor and NF-κB pathway activation
• Source: Chapuy et al., Blood 2016 — https://doi.org/10.1182/blood-2016-07-727230
• Source: Nakamura et al., Nature Communications 2016 — https://doi.org/10.1038/ncomms12173
Despite advances in genomic characterization, the potential influence of Hispanic ethnicity on mutation patterns, treatment response, and survival outcomes remains understudied. Data from multiple malignancies suggest racial and ethnic differences may contribute to disparities in outcomes (Patel et al., Cancer Causes & Control 2020: https://doi.org/10.1007/s10552-020-01341-4), but PCNSL-specific insights are limited.
This University of California Hematologic Malignancies Consortium (UCHMC) study evaluates somatic mutation patterns and clinical outcomes in Hispanic versus non-Hispanic patients with PCNSL, with the goal of identifying potential biological contributors to disease behavior and prognosis across ethnic groups.
Abstract Number: abs25-12597
Title: Ultra high-risk multiple myeloma with early mortality despite quad-class and BCMA-directed therapies: Clinical and molecular insights
Room: OCCC – West Halls B3-B4
Date: Saturday, December 6, 05:30 PM – 07:30 PM EST
Part of session 653. Multiple Myeloma: Clinical and Epidemiological: Poster I
Authors:
Binod Dhakal¹, Maria Hintzke¹, Othman Akhtar¹, Jamila Mammadova², Adetola Kassim², Marcelo Pasquini¹, Meera Mohan¹, Ravi Kishore Narra¹, Sabarinath Radhakrishnan¹, Fumou Sun¹, Anita D’Souza¹, Maher Albitar³, Bhagirathbhai Dholaria²
Affiliations:
¹ Medical College of Wisconsin, Milwaukee, WI, United States
² Vanderbilt University Medical Center, Nashville, TN, United States
³ Genomic Testing Cooperative, Lake Forest, CA, United States
Abstract Body:
Multiple myeloma (MM) outcomes have improved substantially with the incorporation of proteasome inhibitors (PIs), immunomodulatory drugs (IMiDs), anti-CD38 monoclonal antibodies, and B-cell maturation antigen (BCMA)–directed treatments including CAR-T cell therapies and bispecific antibodies (BsAbs). These therapeutic advances have translated into prolonged survival for most patients
• Source: Rajkumar, Blood, 2020 — https://doi.org/10.1182/blood.2019004034
• Source: Costa et al., NEJM, 2021 — https://doi.org/10.1056/NEJMoa2033400
However, a subset of patients exhibits aggressive biological behavior characterized by resistance to all four major therapeutic classes (“quad-class refractory”) and rapid mortality due to uncontrolled disease progression (Gandhi et al., Leukemia 2019: https://doi.org/10.1038/s41375-019-0435-7). Understanding the clinical and molecular characteristics driving this ultra high-risk phenotype is critical for development of risk-adapted treatment strategies and novel therapeutic interventions.
This study examines patients with MM who experienced early death despite exposure to PIs, IMiDs, anti-CD38 monoclonal antibodies, and BCMA-directed therapies. We aim to identify shared clinical characteristics, molecular drivers, and resistance pathways associated with this phenotype, and provide early insights into potential biological mechanisms contributing to therapeutic failure.
Abstract Number: abs25-15523
Title: Real-world validation of the molecular prognostic risk signature in acute myeloid leukemia treated with hypomethylating agents plus BCL-2 inhibitor
Room: OCCC – West Halls B3-B4
Date: Sunday, December 7, 06:00 PM – 08:00 PM EST
Part of session 615. Acute Myeloid Leukemias: Clinical and Epidemiological: Poster II
Authors:
Jonathan Andreadakis¹, Olivia Wilkins², Alexander Shkembi¹, Amy Wu¹, John Basile¹, Anna Krzeczowska³, Alexandra Thalberg⁴, Linda Linderbeck³, Nidia Zapata⁵, Suzane Silbert³, Marci O’Driscoll³, Anne Renteria⁶, Maher Albitar⁷, Lacey Williams⁸, Jason Salemi¹, Amy Alman¹, Tiphaine Martin⁹, David Swoboda³, Gustavo Rivero³
Affiliations:
¹ University of South Florida, College of Public Health, Tampa, FL, United States
² Georgetown University School of Medicine, Washington, DC, United States
³ Tampa General Hospital Cancer Institute, Leukemia Program, Tampa, FL, United States
⁴ University of Minnesota School of Medicine, Minneapolis, MN, United States
⁵ Instituto de Cancerología de la Ciudad de México, Programa de Leucemia, Mexico City, Mexico
⁶ New York University, Leukemia and Bone Marrow Transplantation Programs, New York, NY, United States
⁷ Genomic Testing Cooperative, Lake Forest, CA, United States
⁸ University of North Carolina, Chapel Hill, NC, United States
⁹ Icahn School of Medicine at Mount Sinai, New York, NY, United States
Abstract Body:
Response to hypomethylating agents (HMA) plus BCL-2 inhibitors is a major determinant of outcomes in older adults with acute myeloid leukemia (AML). Döhner et al. identified a four-gene molecular prognostic risk signature (mPRS) — incorporating FLT3, NRAS/KRAS, and TP53 mutations — that stratifies response benefit to HMA + BCL-2 inhibitor therapy into superior, intermediate, and low-benefit groups, with subsequent validation at MD Anderson Cancer Center
• Source: Döhner et al., Blood 2022 — https://doi.org/10.1182/blood.2022016867
To support clinical adoption, real-world validation of mPRS is needed. The primary objective of this study was to assess complete remission (CR) + CR with incomplete recovery (CRi) among elderly AML patients classified by mPRS. A secondary aim evaluated whether immunophenotypic differentiation arrest influences treatment benefit, given known enrichment of FLT3-ITD and RAS mutations in monocytic / granulocytic subtypes.
After IRB approval, 123 AML cases aged ≥60 years treated with HMA + BCL-2 inhibitor were analyzed. Clinical and molecular features were summarized using descriptive statistics; associations with outcomes were assessed using chi-square and t-tests.
Median age was 71 years (range 60–93), and 59.3% were male. ELN 2022 risk distribution was: favorable 11%, intermediate 18%, and adverse 72%. CR+CRi rates trended higher in adverse-risk patients (66%) versus intermediate (25%) and favorable (9%), p=0.09.
Using the four-gene mPRS, CR+CRi rates were significantly different across groups:
• Superior benefit: 43%
• Intermediate benefit: 37.5%
• Low benefit: 19% (p=0.03)
Low-benefit patients were enriched for complex karyotype (91%), whereas normal karyotype predominated in superior (41%) and intermediate (55%) groups (p<.0001 and p=0.008, respectively).
When analyzed by immunophenotype:
• HPC-like AML (less differentiated): superior 56%, intermediate 22%, low 22% (p=0.04)
• Maturing-like AML: superior 31%, intermediate 50%, low 19%
TP53 and myelodysplasia-related mutations (ASXL1, BCOR, RUNX1, splicing factor genes) were more common in HPC-like AML; FLT3-ITD predominated in maturing-like AML (79% vs 21%; p<0.001), with higher N/KRAS in the same group.
Keywords: Diseases; Research
Abstract Number: abs25-4126
Title: Developing an artificial intelligence–based transcriptomic signature to identify HOXA-MEIS1 pathway abnormalities for patient selection in menin inhibitor therapy
Room: OCCC – West Halls B3-B4
Date: Sunday, December 7, 06:00 PM – 08:00 PM EST
Part of session 618. Acute Myeloid Leukemias: Biomarkers and Molecular Markers in Diagnosis and Prognosis: Poster II
Authors:
Maher Albitar¹, Hong Zhang¹, Gustavo Rivero², David Swoboda², Sally Agersborg¹, Adam Albitar¹, Ahmad Charifa¹, Andrew Ip³, Andre Goy³, Andrew Pecora³, David Siegel³, Kelly West Fitzpatrick³, Katherine Linder³, Jamie Koprivnikar³, James McCloskey³
Affiliations:
¹ Georgia Southern University, Statesboro, GA, United States
² Tampa General Hospital Cancer Institute, Tampa, FL, United States
³ John Theurer Cancer Center, Hackensack, NJ, United States
Abstract Body:
The menin–KMT2A (MLL) complex is essential for activation of the HOXA-MEIS1 transcriptional program, which drives leukemogenesis in specific subsets of acute myeloid leukemia (AML). KMT2A rearrangements (KMT2Ar) and associated fusion genes are well-recognized oncogenic drivers in pediatric and adult AML, and targeted menin inhibition represents an established therapeutic approach in these leukemias
• Source: Krivtsov & Armstrong, Blood 2007 — https://doi.org/10.1182/blood-2006-07-001263
• Source: Dzama et al., Cancer Discovery 2020 — https://doi.org/10.1158/2159-8290.CD-19-1323
However, HOXA-MEIS1 pathway activation can occur independent of KMT2Ar (e.g., through NPM1 or DNMT3A mutations), and these patients may also derive clinical benefit from menin inhibitors
• Source: Uckelmann et al., Nature 2020 — https://doi.org/10.1038/s41586-020-2368-1
We hypothesized that KMT2Ar leukemias share a generalized transcriptomic signature reflecting HOXA-MEIS1 activation, and that an artificial intelligence (AI) model trained on this signature could identify KMT2A-negative (KMT2An) AML cases with similar biology.
Using RNA-sequencing datasets from AML patients with KMT2Ar, an AI-derived gene expression signature was developed to characterize HOXA-MEIS1 pathway activation. This model was then applied to KMT2An AML samples to assess for presence or absence of the signature, with the goal of identifying additional populations who may be sensitive to menin-targeted therapy.
This AI-based transcriptomic approach may expand precision therapy selection beyond conventional cytogenetic criteria and refine eligibility algorithms for menin inhibitor treatment.
Keywords: Treatment Considerations; Artificial Intelligence (AI)
Abstract Number: abs25-7855
Title: Artificial intelligence–based transcriptomic signature for diagnosing dark zone lymphoma in patients without MYC gene rearrangement
Room: OCCC – West Halls B3-B4
Date: Sunday, December 7, 06:00 PM – 08:00 PM EST
Part of session: 621. Lymphomas: Translational – Molecular and Genetic: Poster II
Authors:
Andrew Ip¹, Maher Albitar², Hong Zhang³, Sally Agersborg², Adam Albitar², Ahmad Charifa², Andrew Pecora¹, David Siegel¹, Lori Leslie¹, Andre Goy¹, Tatyana Feldman¹
Affiliations:
¹ John Theurer Cancer Center, Hackensack, NJ, United States
² Genomic Testing Cooperative, Lake Forest, CA, United States
³ Georgia Southern University, Statesboro, GA, United States
Abstract Body:
Double-hit diffuse large B-cell lymphoma (DLBCL) and Burkitt lymphoma (BL) are categorized as dark zone lymphomas (DZL) based on shared origin from densely proliferating B cells in germinal center dark zones and distinct transcriptional biology. These entities are associated with poor clinical outcomes and require intensified therapeutic approaches
• Source: Schmitz et al., NEJM 2018 — https://doi.org/10.1056/NEJMoa1801445
• Source: Wright et al., Blood 2020 — https://doi.org/10.1182/blood.2019000937
Unique gene-expression programs distinguish DZL from non-DZ DLBCL, including MYC pathway activation and germinal center–associated signatures. Dose-intensified chemotherapy has improved the prognosis of MYC-rearranged (“double hit”) DLBCL and BL; however, biologically similar non-MYC rearranged DZL cases remain under-recognized and understudied.
We used targeted transcriptomic profiling combined with artificial intelligence (AI) to identify a DZL-specific gene-expression signature. Once trained and validated, the AI model was applied to 187 DLBCL cases lacking MYC rearrangement (DLBCLn) to determine whether they harbored a DZL-like transcriptomic pattern.
This AI-based diagnostic approach aims to improve biological classification of DLBCL beyond MYC status, identify high-risk patients who may benefit from intensified therapy, and enable incorporation of DZL biology into routine clinical decision-making.
Keywords: Artificial Intelligence (AI); Diagnostics; Aggressive Lymphomas
Abstract Number: abs25-12216
Title: Molecular profiling and kinetics of circulating multiple myeloma cells (CMMCs) predict resistance to bispecific antibodies in relapsed and/or refractory multiple myeloma
Room: OCCC – West Halls B3-B4
Date: Sunday, December 7, 06:00 PM – 06:00 PM EST
Part of session 653. Multiple Myeloma: Clinical and Epidemiological: Poster II
Authors:
Binod Dhakal¹, Michael Slade², Mostafa Mohamed³, Mark Schroeder², Ravi Kishore Narra¹, Othman Akhtar¹, Hima Bansal³, Maria Acevedo-Calado³, Oren Pasvolsky³, Mahmoud Gaballa³, Melody Becnel³, Donna Weber³, Krina Patel³, Sheeba Thomas³, Jing Christine Ye³, Meera Mohan¹, Marcelo Pasquini¹, Fumou Sun¹, Keith Stockerl-Goldstein², Anita D’Souza¹, Ravi Vij², Maher Albitar⁴, Robert Orlowski³, Hans Lee⁵
Affiliations:
¹ Medical College of Wisconsin, Milwaukee, WI, United States
² Washington University School of Medicine, St. Louis, MO, United States
³ The University of Texas MD Anderson Cancer Center, Houston, TX, United States
⁴ Genomic Testing Cooperative, Lake Forest, CA, United States
⁵ Sarah Cannon Research Institute, Nashville, TN, United States
Abstract Body:
Bispecific antibodies (BsAbs) directed against B-cell maturation antigen (BCMA) and GPRC5D produce rapid and deep responses in relapsed/refractory multiple myeloma (RRMM) but are limited by variable durability and the emergence of resistance
• Source: Munshi et al., NEJM 2021 — https://doi.org/10.1056/NEJMoa2024850
• Source: Lee et al., Lancet Oncology 2023 — https://doi.org/10.1016/S1470-2045(23)00114-2
Circulating multiple myeloma cells (CMMCs) represent a minimally invasive approach to real-time monitoring of tumor biology and clonal evolution
• Source: Manier et al., Blood 2018 — https://doi.org/10.1182/blood-2017-09-806000
However, early biomarkers that predict resistance to BsAbs remain unmet clinical needs. We evaluated whether CMMC kinetics and molecular characteristics correlate with treatment response to BsAbs in RRMM.
Serial CMMC quantification and molecular profiling were analyzed to determine associations between:
• Baseline CMMC burden
• Early fold-change dynamics post-therapy
• Genomic and transcriptional signatures
and depth/durability of response to BCMA- or GPRC5D-targeted BsAbs.
This study investigates whether early changes in circulating disease burden and mutational/transcriptomic features can serve as predictive biomarkers for BsAb resistance and clinical outcomes.
Keywords: Minimal Residual Disease; Immunotherapy; Liquid Biopsy
Abstract Number: abs25-4127
Title: Artificial intelligence–based transcriptomic signature for identifying IDH1- and IDH2-driven acute leukemia and detection of signature prevalence in mutation-negative cases
Room: OCCC – West Halls B3-B4
Date: Monday, December 8, 06:00 PM – 08:00 PM EST
Part of session 618. Acute Myeloid Leukemias: Biomarkers and Molecular Markers in Diagnosis and Prognosis: Poster III
Authors:
Maher Albitar¹, Hong Zhang², Gustavo Rivero³, David Swoboda³, Sally Agersborg¹, Adam Albitar¹, Ahmad Charifa¹, Andrew Ip⁴, Andre Goy⁴, Andrew Pecora⁴, David Siegel⁴, Kelly West Fitzpatrick⁴, Katherine Linder⁴, Jamie Koprivnikar⁴, James McCloskey⁴
Affiliations:
¹ Genomic Testing Cooperative, Lake Forest, CA, United States
² Georgia Southern University, Statesboro, GA, United States
³ Tampa General Hospital Cancer Institute, Tampa, FL, United States
⁴ John Theurer Cancer Center, Hackensack, NJ, United States
Abstract Body:
Mutations in IDH1 and IDH2 lead to neomorphic enzymatic activity, production of D-2-hydroxyglutarate, and epigenetic dysregulation contributing to leukemogenesis
• Source: Stein & DiNardo, Blood, 2019 — https://doi.org/10.1182/blood-2018-10-844662
The clinical significance of IDH1/2 mutations in AML remains debated, as outcomes may not markedly differ from mutation-negative disease when treated with conventional chemotherapy. However, addition of IDH inhibitors to venetoclax-based or intensive regimens improves outcomes in IDH1/2-mutated AML
• Source: DiNardo et al., NEJM, 2020 — https://doi.org/10.1056/NEJMoa1912913
We hypothesized that IDH1/2-mutant AML harbors a distinct transcriptomic phenotype identifiable by artificial intelligence (AI), and that this molecular signature may exist in a subset of patients without detectable IDH1/2 mutations using standard sequencing.
Using transcriptomic data from IDH1-positive (IDH1p) and IDH2-positive (IDH2p) AML samples, we developed an AI-based signature to characterize IDH-driven biology. This model was then applied to IDH-mutation-negative AML (AMLn) to determine prevalence of IDH-like molecular patterns. Identifying such biology may expand the population eligible for IDH-targeted therapy and refine precision treatment strategies.
Keywords: Artificial Intelligence (AI); Epigenetics; Targeted Therapy
Abstract Number: abs25-7376
Title: Bone marrow microenvironment overlap between VEXAS and myelodysplastic syndrome demonstrated by targeted transcriptomics and artificial intelligence
Room: OCCC – West Halls B3-B4
Date: Monday, December 8, 06:00 PM – 08:00 PM EST
Part of session 636. Myelodysplastic Syndromes: Basic and Translational: Poster III
Authors:
Maher Albitar¹, Sally Agersborg¹, Hong Zhang², Adam Albitar¹, Ahmad Charifa¹, Andrew Pecora³, David Siegel³, Andre Goy³, Jamie Koprivnikar³, James McCloskey³, Katherine Linder³
Affiliations:
¹ Genomic Testing Cooperative, Lake Forest, CA, United States
² Georgia Southern University, Statesboro, GA, United States
³ John Theurer Cancer Center, Hackensack, NJ, United States
Abstract Body:
VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) syndrome is a recently described inflammatory condition driven by somatic UBA1 mutations, characterized by systemic inflammation and cytoplasmic vacuoles in myeloid precursors
• Source: Beck et al., NEJM 2020 — https://doi.org/10.1056/NEJMoa2026834
Inflammation and immune dysregulation are also well-established components of the bone marrow microenvironment in myelodysplastic syndromes (MDS)
• Source: Sallman & List, Blood 2019 — https://doi.org/10.1182/blood-2018-10-844432
We investigated biological overlap between VEXAS and MDS using a targeted transcriptomic approach integrated with artificial intelligence (AI). Bone marrow transcriptomes from VEXAS cases (N=59) were compared with those from patients with MDS (N=1021) and from individuals without hematologic malignancy or with clonal hematopoiesis of indeterminate potential (CHIP) (N=1030).
Our objective was to characterize shared pathways and microenvironmental signatures between VEXAS and MDS, identifying inflammatory drivers and potential diagnostic ambiguities. This AI-driven comparative analysis may support improved understanding of disease biology in both entities and enhance recognition of cases with overlapping features.
Keywords: Bone Marrow Microenvironment; Artificial Intelligence (AI); Inflammation
Thank you to our collaborators, who are listed below:
– Baylor College of Medicine
– City of Hope National Medical Center
– Columbia University Irving Medical Center
– Columbia University Vagelos College of Physicians and Surgeons
– Georgetown University School of Medicine
– Georgia Southern University
– Hackensack Meridian Health
– Icahn School of Medicine at Mount Sinai
– Instituto de Cancerología de la Ciudad de México
– John Theurer Cancer Center at Hackensack University Medical Center
– Medical College of Wisconsin
– New York University
– Pathology Associates
– Sarah Cannon Research Institute
– Tampa General Hospital Cancer Institute
– The University of Texas MD Anderson Cancer Center
– UC Davis Comprehensive Cancer Center
– UC Irvine
– UC San Diego (UCSD)
– UCSF Fresno
– University of Minnesota School of Medicine
– University of North Carolina
– University of South Florida
– Vanderbilt University Medical Center
– Wake Forest University School of Medicine
– Washington University School of Medicine
Read more Articles and News from GTC

GTC opens Houston laboratory, expanding national precision oncology network
New central U.S. location strengthens testing capacity and turnaround times while positioning GTC within the Texas Medical Center ecosystem HOUSTON — March 12, 2026 — Genomic